Assembling your first system of robots
Sight built-in robot collection (last tested 18th August 2002, 17:02)
Reading
complex URLs and using DAS servers
Creating robots for servers that
return XML response
Using the SSH and local programs
Getting
started
Installation
and run
If you already have java
1.4.0 or compatible in your computer, Sight can be installed by cross-platform
installer (executable .jar), tested on Windows and Linux. If you do not
have java or even do not known what it is, use the provided Windows executable
(.exe). This installer is larger and not cross platform, but it installs all
needed components, including java virtual machine. Both installers will leave
"uninstall" option. The last method is to unpack the provided .zip
file into any folder and then install using provided shell scripts.
Assembling
your first system of robots
After you start Sight, you
should see the following dialog window:

This window is used to
assemble a group of robots for a certain data flow. For example, let's suppose
that you have a list of NCBI protein identifiers and would like
- Retrieve the protein sequence for each
identifier.
- Predict the potential location in the cell
and the number of transmembrane helixes for each protein.
The tree on the left
defines the current data flow diagram. New robots can be added to any node by
selecting it and then pressing the top left button ("add existing"). The
robot receives the sub task from the upper node (parent). The top-most node
initiates the work of the application. This is a Sight built-in robot that can
just read identifiers from files, open pop-up dialogs to enter the new values
or read the files containing the sequences in Fasta format.
The first robot you need to
add is fastaReaderNCBI. It is located in the folder impl/sequence_Readers
(another robot in this folder reads protein sequences from Ensembl). After
adding this robot, select it. The pane on the left will show, how the master
robot output will be connected with the fastaReaderNCBI input. This case is simple, and no
editing is required.
Now add the robots PSORT, TargetP
and TmPred, located in the impl/ protein_analysers folder (you can choose
all at once). All these robots takes 'Sequence' as an input, and fastaReaderNCBI provides 'Sequence' as an output. Hence it is still not necessary to
set the connections manually.
Now select TmPred and
click on Web form tab. You will see some additional options that this
internet server (http://www.ch.embnet.org/software/TMPRED_form.html) provides to adapt the analysis to
your specific needs (you can set minimal and maximal segment lengths, for
instance). Even though you can leave the default settings, most of the time
(for example, when selecting the BLAST database) you will need to alter them. For
now, just increase the maximal helix length till 35:

Starting
the assembled system
Your application is now
complete and ready to run. Java programmers can press "generate" to
see and modify the java code, made by Sight to perform this task. They may then
switch into analysing that file instead of reading the rest of this chapter. If
you want just to run the application, press the "run application"
button.
Before start you will be
asked if you want to exit the Sight environment before starting the application
program. For machines with moderate memory resources it is better to close the
current program, avoiding system overload (choose "Close it").
If you choose to restart
the created application later, this can be done by choosing Application|Run existing. Sight will also generate the batch file (myTeam_team.bat by
default), containing the required starting commands. This file will be stored
to the default working directory. If you need to start the application
frequently, rename the this file preventing it from being replaced each time
you start the new application.
ome old versions of
Windows95 may contain a bug, preventing the generated agent system from
starting normally from inside another java application. If the system can be
built, but is not operating as expected, start it manually by launching
myTeam_team.bat. You can also start the application by executing the
command that is shown before the attempt to start. Advanced users may use this
information to launch the application under Linux.
The first thing this
application needs is to get a file with the stored identifiers. Hence it starts
from the file open dialog. Choose prepared example file NCBI_list.txt .
After that, Sight runtime environment appears:

The "Console" tab
contains messages from all working internet robots. The Internet tab allows to
view all data that the program is receiving from the internet (in html). The
initial "Task" tab is for general information.
The set of pictograms below
indicates the current status of each internet session (connected, disconnected,
waiting, retrying and so on). The right mouse button on the icon opens the session
dialog box.
Even though the real Sight
tasks may need weeks to complete, this one should be finished in 5-10 minutes
or even faster. The final result are 3 html files, that must appear inside the
logging folder. The name of the folder depends from where the Sight was
installed. It is printed in the "Task" tab console and accessible by
the created shortcuts. If you still need to locate it manually, look for a /Log
subfolder in the Sight installation directory (may be C:\Sight_polygon\Polygon\Log
or similar). The log files are cross linked and differ in the number of
details, provided on the received server responses. This is what the most
abstract index. htm should look like:
NP_055194 details..
- fastaReaderNCBI results:
found and
processing fastaReaderNCBI result 0: >gi|7657289|ref|NP_055194.1|
potassium channel, subfamily V, member 1; neuronal potassium channel alpha
subunit [Homo sapiens]
- Dimers
results:
found homodimer
0.758
PSORT results:
found 33.3
endoplasmic
found 22.2 plasma
found 22.2 mitochondrial
found 11.1 Golgi
found 11.1 vesicles
- TmPred results:
found 211..230
found 244..265
found 277..295
( ...and so on... )
The report has the
same "nested list" or "tree" structure, as the robot
application you created. Apart results, it contains hyperlinks to the servers,
that actually did the job. In pages under these hyperlinks you may read the
explanations about the algorithm, and also find and cite the
corresponding literature references. Just using web robots is not a
reason to ignore them. The hyperlink under details
lead to more information about this step of analysis.
What if you need to run the
same program again? It was saved to the folder applications and has the
name myTeams_team.java. You can pick it after choosing "Application
® run existing" from menu. You may notice
that the next time it takes much faster for the application to complete your
task. This is because all server responses are cached by default. They are
stored into the folder C:\Sight_polygon\Cache and will expire (by
default) after the 41 day. This folder contains 2 files per robot (the names
correspond to the agent name). These files can be safely deleted, this only
makes the robot use internet for getting the information again. The expiration
time can be altered from the "Robot" tab.
The methods, described in
this chapter, belongs to the most friendly user level. Sight comes with many
pre-defined robots that that can be freely combined into applications. You will
find the complete list of robots in the next page. Most of the tools can be
tunned to your task by changing the settings in "Web form" pane. However
earlier or later this will not be enough. Sight is not just a tool to combine
pre-programmed web services. It is a tool to integrate new web services that
were not even opened at the time of writing this application.
Sequence
manipulation
In some cases it is
necessary to cut a fragment of the known sequence. For example, it may be
interesting to search only for a sequence features that are located at the
certain distance from the other previously found sequence feature, or inside
the other previously found sequence feature. For example, lets test that
PROSITE hits can be found ONLY 40 amino acids upstream the beginning of
segments, predicted by TmPred.
The required application
should look like

Here rangeSelector (that can be inserted by clicking "Subsequence selector"
button accepts the region from TmPred, the sequence from that the region
is selected from fastaReaderNCBI. The Web form tab allows to specify more
details: which subsequence must be selected (left, inside or right from the
specified region) and the maximal length (that allows to say not just
"upstream", but also "40 amino acids upstream"):
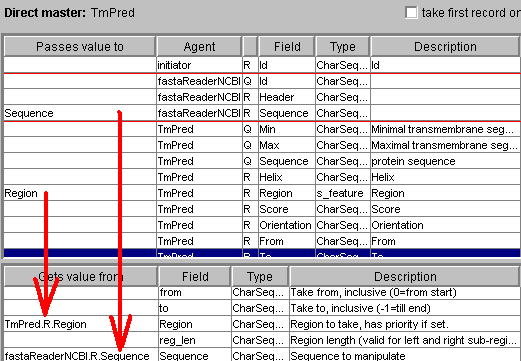
The input of ProSite
must be connected with the output of rangeSelector:
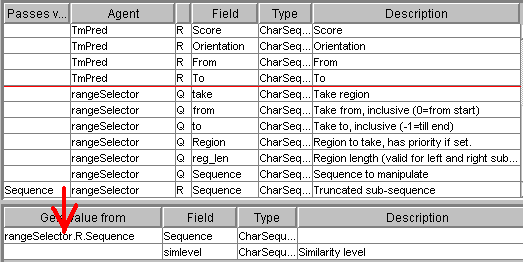
Here is how the output
should look like:
NP_055194
- fastaReaderNCBI results:
found and
processing fastaReaderNCBI result 0: >gi|7657289|ref|NP_055194.1|
potassium channel, subfamily V, member 1; neuronal potassium channel alpha
subunit [Homo sapiens]
- TmPred results:
found and
processing TmPred result 0: 211..230
- rangeSelector results:
found and
processing rangeSelector result 0: left before 211 , length 50
- ProSite results:
found cAMP-
and cGMP-dependent protein kinase phosphorylation site. PS00004
found Protein kinase C phosphorylation site. PS00005
found Casein kinase II phosphorylation site. PS00006
found N-myristoylation site. PS00008
found Microbodies C-terminal targeting signal. PS00342
found and
processing TmPred result 1: 244..265
- rangeSelector results:
found and
processing rangeSelector result 0: left before 244 , length 50
- ProSite results:
found Casein
kinase II phosphorylation site. PS00006
found N-myristoylation site. PS00008
( ... and so on ...)
Filtering
Filtering is used to
perform some steps of analysis only if the certain condition match. For
example, lets suppose we are interested only in ProSite hits, located only inside
the second or third transmembrane helix. We can use the numeric filter
that checks the number-of-helix value in TmPred and transmits to subject robots
only the records where this value is between 2 and 3:

The input of ProSite (the
Sequence field) must be connected with the Sequence field from the output of
fastaReaderNCBI:

The test_value field
of the numberFilter request must be connected with the Helix
field of the TmPred record:

Also, in the Web form tab
of the numberFilter you must set the minimal (2) and maximal (3)
allowed values (Min and Max fields) (this is simple to do and not shown).
Event
listeners
If the purpose of your work
is to detect some specific situation (say the certain similarity search hit
between the first and second transmembrane helixes), and the amount of your
task is huge, it may be difficult to read even the index.htm, not
speaking about the details. For such cases, Sight provides the event listeners.
The event listener agent writes the message to the forth file, Events.htm,
each time it receives control. This message may include up to 4 items from the
requests and responses of the master agent. For example, to report that the
third transmembrane helix was found in a protein, and later that this protein
also contains ProSite hits, the event listeners can be connected in the
following way:

The number filter must be
configured to pass only desired cases (in our situation, the number of
helix=3). The different fields from fastaReaderNCBI and TmPred can be connected
to the fields of SignalMan. Tthe event listener has up to 6 fields to connect
with the different fields of the master robots. The first field of the
SignalMan can also have the fixed value, naming the current agent (all event
listeners write messages to the same Events.htm file). The event listener need
not to be the last node in the tree of analysis. In our case, the output of
numberFilter is also connected to ProSite, and the second event listener
(SignalMan_0) is listening if ProSite returned any hits. The Events.html is
cross linked with index.htm and in our case could look like:
Attention: third transmembrane helix // The value of the first field, fixed, entered in
"Web form" tab.
3 // The value of the second field
Attention: prosite hit found in the protein
with the third transmembrane helix
cAMP- and cGMP-dependent protein kinase phosphorylation site.
Attention: prosite hit found in the protein
with the third transmembrane helix
Protein kinase C phosphorylation site.
If you click on the
hyperlink under notes, you will be brought to the corresponding location in the
index.htm, from where the note was generated.
If there is need to have a
plain text file (for example, for importing into database), this is done by
specifying the text file name the event listener web form. The items in the
text file are not quoted and tab-delimited by default. If needed, the delimiter
and quoting character are also specified from the web form tab.
Saving
Sight applications
You can save and load the
created teams of agents using self-explainable items in File menu. However the
current versions of Java runtime environment does not support versioning of
Swing classes. Hence if you share the saved Sight agent teams, be sure that all
members of you research group are running Sight on the same version of java
runtime environment.
Sight
built-in robot collection (last tested 18th August 2002, 17:02)
|
Function |
File location |
Researcher group
address |
|
Similarity search |
||
|
NCBI nucleotide BLAST |
impl\Blast\blastN_ncbi |
|
|
NCBI protein BLAST |
impl\Blast\blastP_ncbi |
|
|
Ensebl human blast |
impl\Blast\blast_human_ensembl |
|
|
Proteomes and Genomes
Fasta, EBI |
impl\Blast\Ebi_fasta.class |
|
|
Nucleic acid sequence
analysis |
||
|
NCBI electronic PCR |
impl\dna_analysers\E_PCR |
|
|
Genscan web server at MIT |
impl\dna_analysers\geneScanners\GenScan |
|
|
Translates a nucleotide
sequence to a protein sequence, ExPASy |
impl\rna_analysers\rna_translator |
|
|
Protein sequence
analysis |
||
|
QuaternaryStructure
Predictor: ExperimentalHomodimer Classifier |
impl\protein_analysers\Dimers |
|
|
Glycosylphosphatidylinositol
modification site prediction |
impl\protein_analysers\GPI_modification |
|
|
Integrated search in
PROSITE, Pfam, PRINTS and other family and domain databases |
impl\protein_analysers\InterPro |
|
|
Prediction of N-terminal
N-myristoylation |
impl\protein_analysers\Myristoylation |
|
|
Prediction of
mitochondrial targeting sequences |
impl\protein_analysers\Predotar |
|
|
Scans a sequence against
PROSITE (allows mismatches); at PBIL |
impl\protein_analysers\ProSite |
http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_prosite.html |
|
Protein sorting signal
prediction |
impl\protein_analysers\PSORT |
|
|
Simple Modular
Architecture Research Tool; at EMBL |
impl\protein_analysers\SMART |
|
|
Prediction of subcellular
location, at CBS |
impl\protein_analysers\TargetP |
|
|
Prediction of
transmembrane regions and protein Orientation (EMBnet-CH) |
impl\protein_analysers\TmPred |
|
|
NCBI Conserved Domain
Search |
impl\protein_analysers\CDD_ncbi |
|
|
Expression profiling in
silico |
||
|
Finding, where the given
RNA sequence is expressed (uses NCBI database of expressed sequence tags). This
robot depends on the quality and completeness of NCBI EST_human database. |
impl\rna_analysers\whereExpressed |
|
|
Sequence downloaders |
||
|
Reads sequences using
Ensembl identifier |
impl\sequence_readers\fastaReaderEnsembl |
|
|
Reads sequence and fasta
header from NCBI database using given accession number |
impl\sequence_readers\fastaReaderNCBI |
|
Note: The situation in the internet is
constantly changing. Some services may be available unchanged for a several
years, others are constantly under development and you need to generate a new
robot almost monthly. This is why the Sight-like system cannot just contain the
large collection of pre-programmed web robots. It must be able to generate a
new web robots itself. So please do not panic and feel frustrated if five years
after writing this manual the robot you need cannot find the expected data
structure in the pre-programmed place on the web. You will learn how to integrate
almost any web service you currently need in the rest of this
manual.
Integrating
a new web service
Using
tables
Tables in the documents not
just increase readability. If the server response is presented in a well
organised table, Sight or another web robot system usually has no problems with
extracting the informative part. Let's integrate something simple as a start,
for example Expasy PeptideMass tool. PeptideMass cleaves a user-entered protein
sequence with a chosen enzyme, and computes the masses of the generated
peptides. The tool also returns the theoretical isoelectric point and mass
values for the protein of interest. Hence you will see into what pieces your
protein will be cut by known proteases.
First of all, launch your
browser and open http://www.expasy.org/tools/peptide-mass.html, the url where this service is
provided. You should see the submission form. Now, in the Sight main window,
press the "new robot" button. The initial robot creation dialog must
appear:
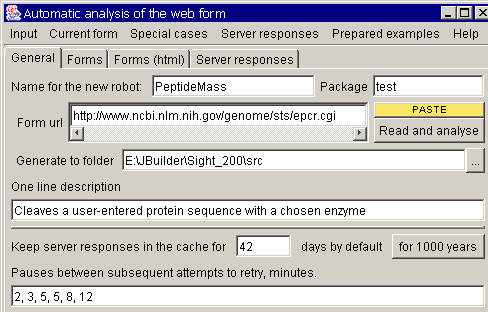
Give your robot a name,
enter a one sentence description, specify package (the sub folder where the
robot will be placed and paste http://www.expasy.org/tools/peptide-mass.html to the "Form url" field. Here
you can also specify, for how long the received server response must be stored
in the cache, and how long the robot should wait before retrying if the server
failed to respond.
Now press "Read and
analyse" and wait for several seconds. Sight must connect the server and
get the code, describing the request form. After it happens, you will see the
form view:

The form elements are
corresponding the same elements in the usual web form that you see in a browser
window. However Sight is displaying more, you may find some extra parameters
that are normally required by the browser. On the right of each parameter you
see two check boxes - para and incl. Para means that this
form parameter will be a variable parameter in request for a finished robot
(currently supported for all types of controls except the check boxes). Incl
means that some value of this field (always default value for
non-parameters) must be included in request for the server. If the server is
responding incorrectly, clearing some Incl boxes can help to get the
desired behavior. By default, button and upload file fields are not included in
requests.
Mand check box appears only for text
areas and means that the field must always be specified and need not have the
default value. This is typically true for protein and nucleotide sequences that
are large and cannot be "default" anyway.
Also, we must provide the
sample protein sequence. Sight has as a set of prepared typical bioinformatical
data that can be used as parameters. Choose "protein sequence" to get
the protein sequence of the human potassium channel KV8.1 into the clipboard. Paste
this into the "protein" section and press "submit".
After getting the response
from the server, the dialog box should appear, where you will be asked to
choose the strategy of analysis. Strategies are ordered by complexity, the most
simple first. To choose the strategy, submit the same sequence as request in
your internet browser. In response, you will see that informative data are
presented in the html table. Between the possible ways of representing the
server response, this one can be the most easily understood by machine. Hence
choose Table then.
In parallel, paste the same
protein sequence in your browser and also press "submit".
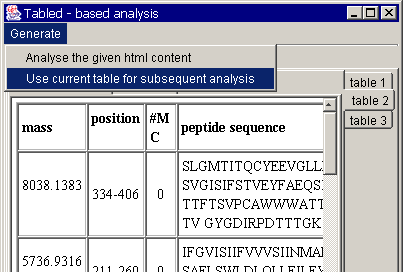
A typical response usually
contains several tables. You will easily see that the required informative part
is presented in table 2. Choose the table 2 for the subsequent analysis. You
will see the following window:

Give the name for each
column, set that the first row must be ignored and check "no html"
boxes to remove html tags from response.
Not all columns are equally
informative. Also, some columns (usually sequences) can be too long for
displaying in the standard report. It is possible to set the report lever for
each field. Set reduced report level for "peptide".
After you finish, press
"Create robot" button. You will see a code of finished the java class
(java code fragment that can be easily inserted into different applications). Close
that window and the class will be saved to the earlier specified destination. Now
you can used the new robot in the same way as any other. For example, you can
combine the chain from NCBI sequence retriever, our new agent PeptideMass
and i.e. transmembrane helix predictor that will predict transmembrane helixes
in the each peptide that remained after digesting the initial protein with
selected the protease.
However in this case you
must specify how the the response from fastaReaderNCBI sequence must be stored to PeptideMass
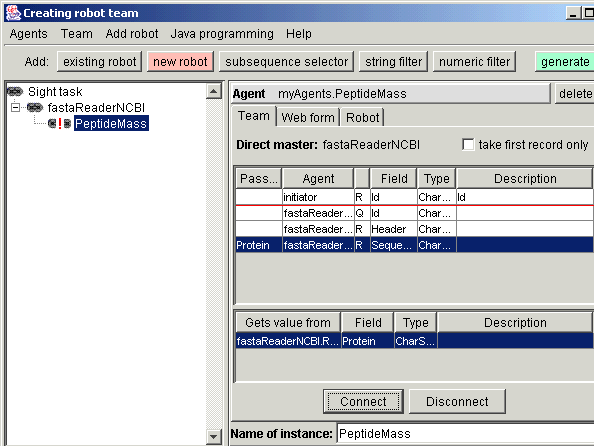
request. In the Team tab you see all fields that fastaReaderNCBI record contains. From them, the Sequence field must be transferred to
the Protein field of PeptideMass request. Just select source, then
destination and press "Connect".
Click now on "Web
form" tab of this window. In this tab, you can change all values that you
declared as parameters. For example, increase Minmass (minimal peptide mass) to 1500.
After you add the next
robot (for example, TmPred), you need to show, how to create TmPred
request from PeptideMass response record. Connect them as it
is shown in the picture below. As a rule, fields for request are taken from the
fields of the immediate master. However, if needed, you can also take them from
the master of the master (fastaReaderNCBI), or from other masters up till the task
initiator.

After you finish, press
"run application" to start the execution process.
Simple direct
URL reading
In some cases the information can be obtained by reading the url part of that is the parameters, required to pass to the server (for example, http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?txt=on&val=NP_055194). This happens when the group that runs the web server releases the corresponding documentation. To create this group of robots, choose Special robots|No form just read url. Sight will create the imaginary form (with get method) for reading the known urls.
Frequently the url consist of constant prefix (in the former case, http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?txt=on&val=), the single variable identifier (in the former case, NP_055194) and may also have the constant suffix. This syntax is usually sufficient to retrieve the database record by its accession number. For easier orientation, this dialog has several built-in examples of the popular records. The simple version of the direct url reading is accessible from the submenu "Basic".
Reading complex URLs and using DAS servers
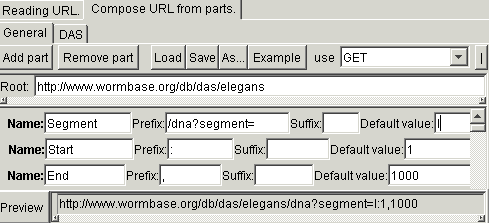
The distributed annotation system (DAS) is a client-server system, where requests are submitted by sending complex URL and responses are returned in XML format. In this case, the URL consists of many variable parts, defining various aspects about the request. It is not following the commonly accepted syntax of the web form. For example, to get a DNA fragment form the chromosome I of the worm C.elegans, it is possible to use URL http://www.wormbase.org/db/das/elegans/dna?segment=I:1,100. For forming such URLs, you must use the more sophisticated generator, accessible from the submenu "Expert". In this generator, the URL consist of the constant base and any number of additional parts. Each part consist of constant prefix, variable identified and constant suffix. The parts also have names that will correspond the names of the request fields in the agent being generated. For the previous example, the form must be filled in the following way:
Some other servers (for example, NCBI) may need a different, but also complicated url syntax. With this advanced generator you have enough freedom to follow various schemas.
The DAS servers follow certain standards, facilitating the integration of the new service. The server can list the annotated features of the given DNA segment, or return the sequence of this segment. The client must specify the region of interest in the segment and the many cases the segments are representing the complete chromosomes. For example, the Wormbase server, mentioned above, allows the following segments:
| Segment name | Valid sequence interval |
| I | [1..15080471] |
| II | [1..15279300] |
| III | [1..13783268] |
| IV | [1..17493790] |
| V | [1..20922239] |
| X | [1..17705013] |
To get information about the available resources (the same server may provide information about several organisms), the valid names of segments and the valid intervals for the sequence, you must switch to "DAS" tab. In this tab, you can specify the address of the DAS server you know (for the list of available servers, see http://www.biodas.org and http://www.tigr.org/tdb/DAS/das_server_list.html). This dialog contains to buttons to get the information about the available data sources and later the segment information for the given source. The new web robot can be specialised to get the sequences or the sequence annotations (create two robots in you need both features). It accepts the name and interval of the segment the request parameters. The response of the first test submission will be directed to the standard Sight agent generator mechanism. For the next step, we recommend to select XML based agent generator or manual specification of landmarks.
Using
Stalker algorithm
Stalker algorithm gets
needed information that is not presented in the tables. It is implemented as
described in (Muslea,I; Minton,S; Knoblock,C.A, 2001). During generation process you need
to mark the informative items in one or several responses. Lets try to get the
items from the first page of NCBI database search results. For this, open the
page http://www.ncbi.nlm.nih.gov/ to look at the search form. Now
start Sight again, press the "New robot" and paste this URL into the
"Form url" field. Press "submit". This page currently
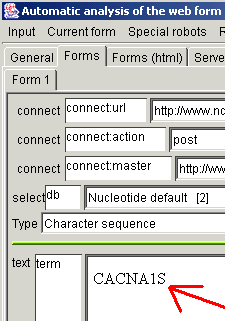
contains just one form. Type something sensible (for example,
"CACNA1S") in the text field near the "term" (in the Sight
web form page).
After you press Submit and
the method selection dialog appears, select Stalker algorithm. After
several seconds you should see something similar as you see in the web browser.
In the displayed response, mark the informative items. If the data are
organized into several records, each containing the same items (as in this
case), the different items in the same record must be marked in different
colours. For marking, select the item and press one of the marking buttons
above:
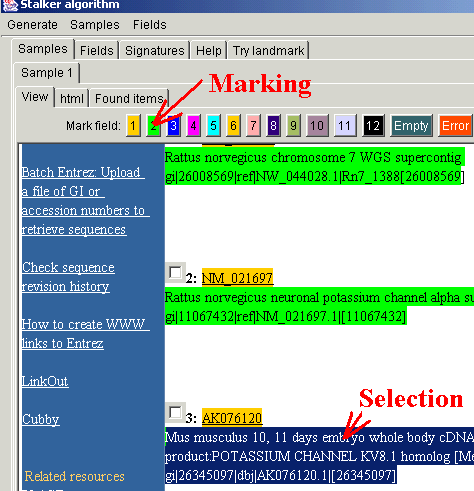
(here, accession number is
marked in orange and record header in green). The algorithm will work much
faster if you mark all information in the page. If the page contains too many
records to mark, try to choose the query that give less hits. The most
important is to mark all items in the first record, the last
record and at least in one in the middle. After you do, choose
Generate|Find landmarks now. For more complex documents the search of landmarks
may take several minutes. After it is finished, in the tab "Found
items" you see the found data. Check this page for false positives and
false negatives.
To ensure that the found
landmarks are suitable for all documents, switch to the form page and enter
something different (like CACNA1A) in the form. The "Sample 2" tab
will appear in the Stalker algorithm pane. Mark the check box "Use the
entire page for testing only" to exclude this page from the landmark
search algorithm (in alternative case, you can use several marked pages a a
training set).
This algorithm
needs the sufficient number of examples. You should also pay the deserved attention for
testing. If you notice, that in the test case, there are false positives or
false negatives in the Found items tab, this means that your need to submit
more requests (they will appear as Sample 3, Sample 4, etc in the tab) and mark
all items. Then try testing again. Usually after several attempts the system
finds the correct logic of the document. To be absolutely sure, you may look at
the "Signatures" tab what was chosen to identify the items.
The "Fields"
section and the rest of the generation process is not much different from the
previous generator.
Manual marking of landmarks
This method is used when
the Stalker algorithm fails, or if you think you can faster and easier specify
the landmarks for the items yourself. This needs skills and some understanding
of html, but may also be more reliable. It may be also preferred when the
server responses contain very many items and it is tedious to mark them all.
An example of such a case
can be the Toulouse BLAST search server, located in France. It is located at http://genopole.toulouse.inra.fr/blast/blast.html . This is a real and rather
difficult example, and quite many important parameters must be set before we
can build a robot for this service. Let's create this robot.
The form here is a little
more complicated, but it contains nothing that deserves special additional
explanation. Many BLAST servers (this server included) are rather sensitive to
changing of the default comparison matrix. To avoid possible error messages,
check the MAT_PARAM param as not includable (clear its incl check box). Select
blasp program by default (this can be changed later) and i.e. trembl
database. Provide protein sequence and submit the request.
The output of this server
is not in a table format, and selecting the table search strategy will not be
helpful. Choose Manual method instead.
The generator screen should
display the complete content of the received html response. Look at the same
response in your browser, submitting the request in paralel. It should contain
3 sections:
- Graphical representation of hits.
- Summary table. This can be used for
automated analysis, but the hit headers here are truncated.
- Complete explanation of the found hits. We
will use this section for the automated analysis.
First of all, we must find,
where in a server response this third section begins. We must show this in a
"raw" html response, not in a view that we normally see in a browser.
Actually html is even better for this task, because we can use some html tags
for orientation where just a whitespace would be displayed in a browser.
Below
the important part of this response is displayed.
(Q9JJ60) BRAIN CDNA, CLONE
MNCB-7013, SIMILAR TO MUS MUS..S= 199 E=3e-50"' ONMOUSEOUT='document.BLASTFORM.defline.value="Mouse-over
to show defline and scores. Click to show alignments"' >
</map>
<CENTER>
<IMG WIDTH=557 HEIGHT=451 USEMAP=#img_map BORDER=1
SRC="nph-viewgif.cgi?180428938330809.gif" ISMAP></CENTER>
<HR>
<PRE>
<PRE>
Score E
Sequences producing significant alignments: (bits) Value
Q9GKU7 (Q9GKU7) HYPOTHETICAL
56.3 KDA PROTEIN. <a href = #Q9GKU7>860</a> 0.0
Q9UHJ4 (Q9UHJ4) NEURONAL POTASSIUM CHANNEL ALPHA SUBUNIT (POTASS... <a href
= #Q9UHJ4>860</a> 0.0
Q9CZR1 (Q9CZR1) 2700023A03RIK PROTEIN. <a href = #Q9CZR1>835</a>
0.0
Q60565 (Q60565) POTASSIUM CHANNEL KV8.1. <a href = #Q60565>835</a>
0.0
P97557 (P97557) NEURONAL POTASSIUM CHANNEL. <a href =
#P97557>832</a> 0.0
Q9BXD3 (Q9BXD3) POTASSIUM VOLTAGE-GATED CHANNEL, SHAB-RELATED SU... <a href = #Q9BXD3>336</a> 2e-91
Q61923 (Q61923) MURINE POTASSIUM CHANNEL PROTEIN. <a href =
#Q61923>150</a> 2e-35
Q18351 (Q18351) C32C4.1 PROTEIN. <a href =
#Q18351>149</a> 5e-35
Q9BYS4 (Q9BYS4) POTASSIUM VOLTAGE-GATED CHANNEL, SHAKER-RELATED ... <a href = #Q9BYS4>146</a> 2e-34
O70259 (O70259) VOLTAGE-GATED POTASSIUM CHANNEL KV1.7. <a href =
#O70259>144</a> 1e-33
</PRE>
<PRE>
><a name =
Q9GKU7></a>Q9GKU7 (Q9GKU7) HYPOTHETICAL 56.3 KDA PROTEIN.
Length = 500
Score = 860 bits (2221), Expect = 0.0
Identities = 437/500 (87%), Positives = 437/500 (87%)
Query: 1
MPSSGRAXXXXXXXXXXXXXXXXXVFCSEGEGEPLALGDCFTVNVGGSRFVLSQQALSCF 60
You
may see that the third section (details) can be locater by finding the Sequences
producing significant alignments sentence and then moving forward till the
string <pre>, html tag defining the start of the preformatted text
section. These must be marked as "far start" and "start":
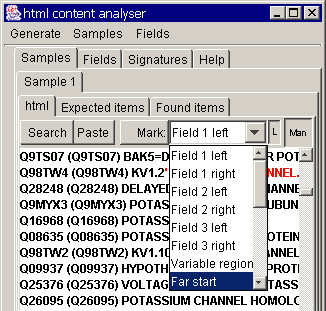
To mark, first select the
region, choose the marking option in the drop-down box above and press the
"mark" button.
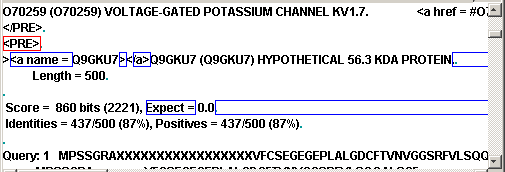
Then
you must mark the left and right signatures for all fields you wish to extract.
There is no universal solution here. However it is possible to mark them in
this way:
|
Field |
Left signature |
Right signature |
Value |
|
1, identifier |
<a name = |
> |
Q9GKU7 |
|
2, header |
/a> |
End of line |
Q9GKU7 (Q9GKU7)
HYPOTHETICAL 56.3 KDA PROTEIN |
|
3, E value |
Expect = |
End of line |
0.0 |
End of line is marked by
placing the cursor anywhere on the line the end of what you need to mark, then
pressing the key END, then holding the SHIFT and pressing ARROW DOWN and HOME. Here
is the fragment of the window with marked regions:
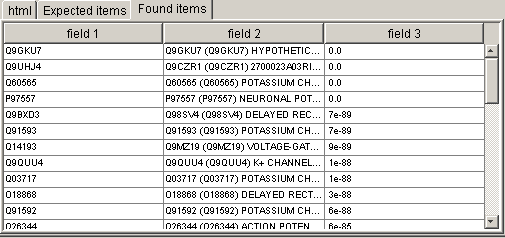
Now we can test our
signature combination. Choose Generate|Try current
settings from the
window menu. Then switch to the "Found items" section. You will see
the table with all found fields:
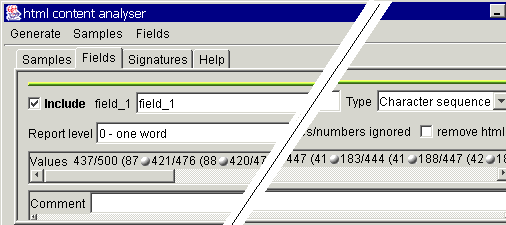
Switch now to the Fields
tab. Here you will see similar information as for the table based robot fields.
It is also recommended to give the fields meaningful names and provide some
comment.
Now we could create the
robot, but it may not be very reliable. In some cases, BLAST responds
differently. What about the possible errors? To get an example of error
message, switch back to the form window (do not close the current one) and
submit the empty sequence. You will see the following output:

"Short error
description" will the most probably appear after most of the errors. Mark
this as "Error" using the same marking controls as for the field
boundary.
But that if no significant
similarity is found? This is perfectly ok and should be treated as an empty
result, not as an error. To see the example of response for this message, just
submit "aaaaaaaaaaaaaaaaaaa" as the sequence. You will see, that in
this case the message " ***** No hits found ****** " appears. Mark it
as "No result" case.
You can mark several
"error" and "no result" cases in different responses.
Now, for the final test,
just submit some another sequence (take a piece of CACNA1A from http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?view=fasta&val=6166047). You will get a different
response. Choose "try current settings" again to see if the robot is
able to understand a completely different response. If it finds all expected
items, the work can be finished by generating this robot and using it later.
"No results" and
"Error" cases can also be marked in the "Server responses"
tab of the main form window. In this way, they can be detected independently
from the used way of analysis (plain text, xml and table based analysers have
no built-in detection for these situations). However, for the signature based
analysis, it may be more convenient to do all markings in a single window.
As with Stalker algorithm,
you can make subsequent submissions and check if your landmarks are suitable
for the other documents.
Using
plain text analysers
Many older servers provide
the informative part of response in the form of "plain text" tables. In
this format, the items are just separated by spaces and "end of line"
symbols. This way of output is preferred by many groups that are concentrating
on a quality of the method, not on creating an impressive decoreative ouptut. Integrating
such responses may be easier than it may look. Let's integrate "Prediction
of transmembrane regions in prokaryotes using the Dense Alignment Surface
method (Stockholm University)", located at http://www.sbc.su.se/~miklos/DAS/ .
The form is very simple
here, containing just a sequence field and one button. Paste the protein
sequence in the sequence field. After the "Choose method" dialog
appears, choose the Plain text method. The response contains a required
table that is displayed in a simple way, without any html formatting commands
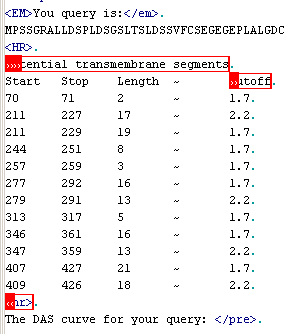
The string "Potential
transmembrane segments" just before the table can be used as "Far
start" marker. This means that the table is below these words, but some
text may still preceed the informative items (in this case, the table header
preceeds informative items). To indicate the start of the table itself, mark
the last work in the header ("Cutoff") as the "Start from".
To show where the table ends, mark the html tag <hr> (is shown as
horizontal line in a browser) as the "end after". Choose "Try
current settings" from menu now and look at the result:
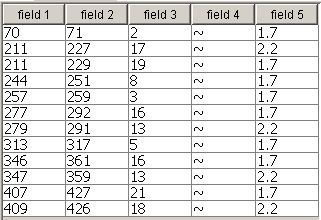
Field 4 with decorative
element can be excluded. Other fields contain different parameters of the
predicted transmembrane segments. Naming fields and setting they options is not
different from the signature based method. Empty result and error cases must be
selected in the main form "responses" tab.
FASTA format. Apart from tables, a frequent type
of responses is fasta format. In this format, each record starts by the symbol
'>' followed by header. The header is terminated by the 'end of line'
symbol. Then one or several lines of the sequence follows. To use plain fasta
format, you must just check the "Fasta format" option in a "Data
reader" section. If the section with this format, is flanked by parts of a
response with a different (usually html) structure, you need to mark
"start from", "end after" and possibly "far
start" regions.
Using
redirectors
The server and your
computer can stay connected just for a limited abount of time. After you submit
a request, the server must reply in 10-20 minutes. If the response is not ready
after this time, the connection will be broken anyway, displaying just an error
message. However some bioinformatical computations may take far longer time to
complete.
This problem is solved by
changing a single connection by several. The first connection is just to submit
a request. The response is usually a hyperlink to the stored results. These
results, however, are not ready at the moment. The hyperlink must be visited
after a certain time, but not immediately. If the results are still not ready,
the server usually just resends the same page, as after the submission of
request.
Sight is able to recognise
many such cases without your help. These includes standard html redirection
tags and the most simple javaScript constructions. However, the robot may
overlook some complicated cases. In this situation, you might need to configure
the redirection mechanism manually.
To make the Sight system
follow the link in a received response, just mark left and right borders of the
hyperlink to follow in the "Server responses" tab that you can also
use to mark the agent - independent "zero" and "error"
cases:
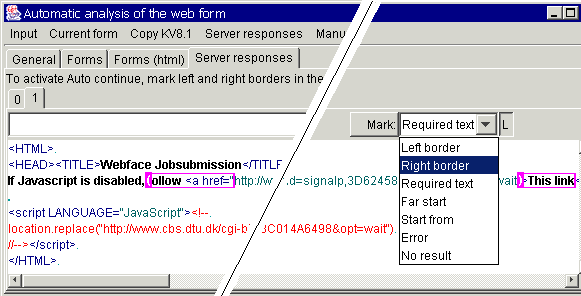
If the response contains
both signatures, the string between them is used as a hyperlink that must be
followed after 2 minutes. If the page hidden under the hyperlink still contains
both signatures, it is rechecked again (also after 2 minutes). I this way, the
server is checked until results are ready. If it is difficult to find the
hyperlink just by specifying the borders, you can also mark far start and start
for the area where the hyperlink is expected.
Creating
robots for servers that return XML response
XML documents can be returned by some web servers
instead of html documents. The typical XML document contains a lot of
information, organized in the same way as files in computer (folders and
subfolders). Even more, when two folders with exactly the same name are not
allowed, the two (or much more) XML entries with identical names are frequent,
for example, in NCBI documents. All this makes extracting the needed
informative part not so trivial. As XML document contains many nested
subfolders, and Sight currently only supports one nesting level (records in
response), the document must be first transformed into the simpler data
structure, containing only one nesting level. Sight uses XSLT transform language to perform this
transformation. This is near complete programming language, and Sight uses the
specialised code generator to create a transform definition. If needed, you can
later view and change the generated transform file.
The example of xml response
can be obtained using direct url reading form (see above) and choosing the
pre-programmed built in url "NCBI XML". In the displayed xml
response, you must mark the following items:
1. The main group. This
indicates the xml folder that will correspond the Sight record. As the Sight
robot response can have multiple records, it can be (and often are) several
main groups in the document. The main group is specified by selecting the
"group" radio option while the wanted folder is selected. If the main
group is not selected, the document root is supposed (not recommended).
2. The items. All items
must be inside the main group. They correspond the fields in the Sight record. If
no other information is specified, and the main group has several items with
the same name and identical subpath, the first such item will be taken:

After you press
"Generate and try transform", the transform will be generated (you
can see and modify it in the XSLT tab) and the transformed document will be
shown (in "Transformed document" as a tree and in "Output"
as a text). The transformed document must contain one root
("<Sight>") with the multiple folders, all having the same name
<Sight_record>. The <Sight_record's> must contain the named
folders, each having the text content. The names of these folders will be the
names of the fields (duplicates are not allowed), and the text content will be
the values for the fields. The field definitions will be created after choosing
Fields|Create fields and can be modified in the Fields tab:
<?xml version="1.0" encoding="UTF-8"?>
<Sight-record>
<Seqdesc_comment>
Summary:
L type voltage-dependent calcium channel controls release of calcium in
skeletal muscle cells. Alpha 1 (CACNA1S) is one of the five subunits that
constitute this calcium channel. Mutation in CACNA1S has been reported to
associate with malignant hyperthermia susceptibility and hypokalemic periodic
paralysis.
</Seqdesc_comment>
<Org-ref_taxname_t>Homo
sapiens</Org-ref_taxname_t>
<Org-ref_common_t>human</Org-ref_common_t>
<Object-id_id_t>9606</Object-id_id_t>
</Sight-record>
<Sight-record> (...)
This is simple, but in many cases you need to locate information from the
folders having the same names. In such case the correct folder can only be
determined by the presence of the certain agreed value ("anchor"). This
agreed value can be stored in the attribute of the text field. It can be
present immediately in the folder we need to select or in some subfolder of
such folder. To locate such information, you need to use the second level
groups and anchors. Lets see the following example (only two small parts of the
document are shown). The main group was marked at the start of the document:
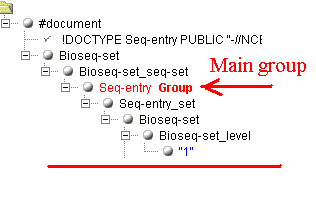
Now, we need to get the mapping information of the sequence
("1q32"). How to do this when there are several subfolders
"SubSource", and the needed folder can only be identified by the
value "map" of the attribute "value" in the folder
"SubSource_subtype"?
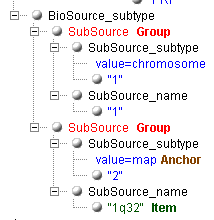
If this case, you need to mark the second level groups. In the group you
need, you mast mark the indicating item as an "Anchor", and the
informative item as "Item". The generator will create the xml
transform, containing among other information the tag <xsl:if
test="SubSource_subtype/@value='map'">. It will extract correctly
the value "1q32" and not "1" from the previous folder. Sight
supports any number the second level groups. Each such group must have its
anchor defined and normally contains at least one informative item to extract. If
the items in a different second level groups have identical names, numbers are
appended to make them different.
Using
Weka classifiers
Weka is a package of tools that, among other
things, can be used to classify the text documents into two (or more) groups. For
example, you may need to classify FASTA sequences into the proteins, belonging
or not belonging to the certain group, using the sequence header field. Weka
comes with a large number of classifiers. Sight allows you to try all of them
automatically, and choose the most suitable one. Before creating robot that
uses Weka classifier, you need to have:
- The list of keywords. This is the list of
specific words that will be minded in the documents being
classified.
- The list of positive examples that fit
your search criteria.
- The list of negative examples that does
not fit your search criteria and must be rejected.
- The testing sets of positive and negative
examples. They will be used to test the reliability of the generated
classifier.
For you first try you can
use the provided set of keywords and two lists (proteins - sodium channels and
proteins - potassium channels). To go to Weka robot generator, choose this
option from "Special robots" in the "New robot" window.
In Weka window, you can
plug-in any later Weka version (you can find them in SourceForge). Sight comes with Weka 3.3.4, where our
development group corrected some minor serialization bugs. After you provide
keyword and training sets, you can test a selected classifier (select from the
combo box below and press "start training") or compare all classifiers
(just press "Compare classifiers"). If you compare all classifiers,
Sight will show you the table, where they are sorted by reliability (for your
training set and your testing data) and then by the working speed. If you do
not specify the testing sets in the "Testing set" tab, the training
data will be also used for testing (not recommended for work, just for
learning).
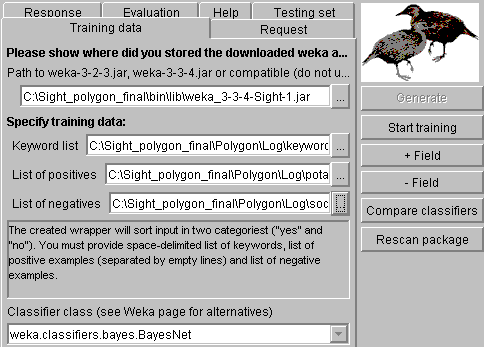
The beginning comparison
table should look like:
Classifier class Errors, Duration (classification/training), notes
weka.classifiers.trees.Id3 , 0 , 30/221 , (tp 16, tn 15, fp 0, fn 0)weka.classifiers.functions.Logistic , 0 , 30/4066 , (tp 16, tn 15, fp 0, fn 0)weka.classifiers.bayes.NaiveBayesUpdateable , 0 , 40/161 , (tp 16, tn 15, fp 0, fn 0)weka.classifiers.lazy.IB1 , 0 , 40/180 , (tp 16, tn 15, fp 0, fn 0)weka.classifiers.meta.LogitBoost , 0 , 40/511 , (tp 16, tn 15, fp 0, fn 0)weka.classifiers.rules.DecisionTable , 0 , 41/550 , (tp 16, tn 15, fp 0, fn 0)
Hence for
our example the Id3 classifier seems the most effective. As the rest of
classifiers in the table, it made no errors and also has the shortest
classification duration (30 ms). The next classifier (Logistic) was equally
fast during classification, but it took much more time train it. Now select weka.classifiers.trees.Id3
from the box below and
press "Start training". After the training is
finished, the "Generate" button becomes available -
the robot can be created.
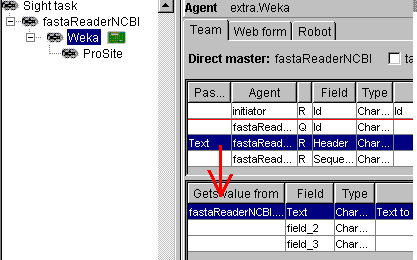
In Sight system, the weka
classifiers work like filters. Below you see how the generate Weka robot
is connected to the NCBI sequence reader: the Header field of the NCBI reader
must be linked with the Text field of the Weka
analyser.
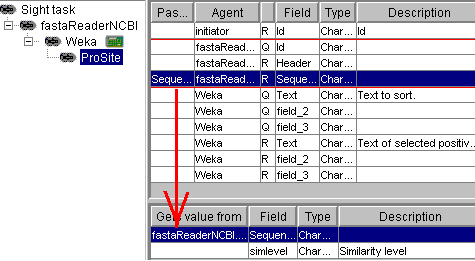
Now, in the second stage,
the ProSite robot is connected as a slave for Weka. It does not take any fields from Weka, but
it takes the Sequence field from the master of its master (sequence
reader NCBI). As a result, ProSite only analyses the records that have
passed the Weka filter:
The results should look
like:
NP_055194 details..
- fastaReaderNCBI results:
found and
processing fastaReaderNCBI result 0: >gi|7657289|ref|NP_055194.1|
potassium channel, subfamily V, member 1; neuronal potassium channel alpha
subunit [Homo sapiens]
- Weka results:
found and
processing Weka result 0: >gi|7657289|ref|NP_055194.1|
potassium channel, subfamily V, member 1; neuronal potassium channel alpha
subunit [Homo sapiens] null null
- ProSite results:
found cAMP-
and cGMP-dependent protein kinase phosphorylation site. PS00004
found Protein kinase C phosphorylation site. PS00005
found Casein kinase II phosphorylation site. PS00006
found N-myristoylation site. PS00008
Using
the SSH and local programs
In addition to web
resources, you can also integrate locally installed programs. Many such
programs are created for Linux, so you may need to install and use Sight under
this operating system.
The program can be also
accessible on another machine that supports SSH protocol. On the most of the
servers this protocol currently replaced TelNet. SSH feature may be needed if
you need to connect more powerful machine or if the applications you need to
integrate run on a different platforms.
Sight starts such program
by executing the given command line. After the name of the executable, the
command line contains arbitrary number of parameters. Each parameter consist of
parameter name, followed by parameter value. Some values (for example, DNA or
protein sequences) are too long for specifying in command line. In this case,
the program requires to save the value in some file and pass the file name
after the parameter name. Sight supports this way of passing the parameters.
The program results are
usually printed to console. Sight captures the program output to console and
then works with it in the same way as it does with the web server responses.
The local program integration
tool is accessible from Special robots|Create
specialised robot --> Execute (SSH or local menu in New
robot window.
Differently from the web
forms, you must specify all parameters yourself. See the program documentation
for their names. The parameter names usually start with dash (like -input) or
slash (like /xml). Specify path to the executable in the local
file system:
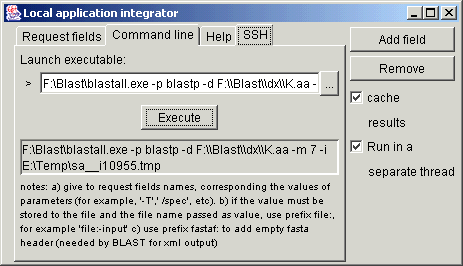
If the value of the
parameter must be written to file, then passing the name of the file in the
command line arguments, the name of such parameter must have prefix file:
(for example file:-i for the sequence input in blastall).
In some cases it is needed to write this file in FASTA format, adding empty
fasta header. In this case the prefix fastaf: is used (for example fastaf:-i
for blastall works better if the xml output is desired).
If you connect via SSH, you
must specify the name of the server on a separate "SSH" tab. The
parameters that must be passed as files will be uploaded to the server using
secure ftp (sftp) protocol. The password and username will be prompted each
time you start the application (not each time you submit a request). If needed,
you may specify prolog and epilog of the SSH session.
After you execute the
selected program first time, the method selection dialog appears. The rest of
the generation process is not different from the generation of the web-based
robot.
If you want to integrate
your algorithm that you wrote in java, you can try Special robots|Create specialised robot --> Java module. In this case, you need to define
not only request fields, but also response record fields. The generated java
skeleton is easy to complete. More details will be available in programmers
guide.
Using
BioQuery
BioQuery (http://www.bioquery.org ) is an advanced system for
creating and submitting queries to NCBI databases. BioQuery saves the created
query file in xml (.qry) format. Sight can read this file, replace any chosen
query terms by Sight variables (request fields) and create the coresponding web
robot. The BioQuery returns the raw server responses that can be automatically
redirected to any Sight analyser (Stalker algorithm, XML transforms and so on).
The provided BioQuery plug-in contains all original GUI classes. So you can
launch the BioQuery application from the Sight environment by pressing a
button. BioQuery plugin is also available from Special robots|Create specialised robot --> BioQuery.
BioQuery provides as free
server-side support that Sight does not need. However you can register to this
service and use it, if you want. You can also download and
use the separate standalone version of BioQuery. The methods to access the
databases are stored in the file Generators/META-INF/querys.xml. You may try to
replace this file by the newer version after s such is released by the BioQuery
development group (backup the old version for peace of mind!).
The BioQuery environment is
very clear, self-explanatory and contains the build in tutorials, so we will
not stop on it. After you create and test a query, save it locally (not on the
BioQuery server), close the BioQuery window and switch back to Sight. In Sight
BioQuery agent generator, select the saved file. The terms will appear in the
form of check boxes. Check the boxes for terms that must be converted to
parameters. After that, press the "Submit" button. After getting the
response to the query Sight opens the dialog for choosing the method to analyse
the BioQuery response.
BioQuery is the the first
plug-in that works independently from the rest of the Sight. In future it is
planned to release more such independent plug-ins. It will be possible to
install them without re-downloading and re-installing the rest of the Sight
application.