These applications were created combining Sight generated agents and custom java code as described in programmers manual
Application 1: Genome walker
Using the Sight modules above, we created a genome walker for analyzing unknown genomic regions. The initial data for the system was a list of clone accession numbers. The sequence retriever robot loaded the sequences from the internet. In the next step, the GenScan module predicted genes. After getting the protein sequence, the following set of robots started their work in parallel: BLAST similarity search (at NCBI), prediction of the transmembrane helixes (TmPred), prediction of the rapid degradation signals (PSORT) and integrated classification (InterPro or PROSITE). The predicted RNA sequence was submitted to the BLAST similarity search using the complete EST database. The list of hits returned by this search contained hyperlinks to the corresponding internet pages. These links were visited to obtain RNA expression patterns. This system generated three levels of reports: a condensed table, an intermediate listing, and a detailed listing that was mainly used for verifying interesting results. The system also detected which NCBI contig was assembled using a clone with a given accession number and included the hyperlink to this contig in the reports. To test this system, we scanned one randomly chosen human chromosome. The list of clones was obtained from UCSC genome assembly 6 (Kent et al., 2002 ). Results are summarized in table 1.
Table 1. Results of scanning of the human chromosome 8 by the Sight based genome walker.
|
Total number of: |
Predicted transmembrane segments (TmPred) |
||||||
|
Clones |
1171 |
None |
830 |
4 |
21 |
||
|
Contigs |
1068 |
1 |
282 |
5 |
11 |
||
|
Clones not in contigs |
103 |
2 |
100 |
6 |
9 |
||
|
Genes, all |
7710 |
3 |
42 |
> 6 |
10 |
||
|
Genes, p>90% |
1305 |
||||||
|
PROSITE profiles |
|||||||
|
Protein sorting signals (PSORT) |
|||||||
|
nuclear |
794 |
Bipartite nuclear localization signal. |
108 |
||||
|
cytoplasmic |
330 |
Various zinc fingers (10 profiles) |
65 |
||||
|
mitochondrial |
66 |
PHD-finger (DNA binding) |
8 |
||||
|
endoplasmic reticulum |
45 |
Ankyrin repeats (2 profiles) |
16 |
||||
|
plasma membrane |
37 |
CUB domain profile. |
14 |
||||
|
extracellular |
27 |
Protein kinases |
13 |
||||
|
cytoskeletal |
2 |
Krueppel-associated box (KRAB) profile. |
12 |
||||
|
Golgi |
2 |
G-proteins (4 profiles) |
10 |
||||
|
vacuolar |
1 |
IG-like domain profile. |
8 |
||||
|
secretory system |
1 |
Other (114 profiles) |
222 |
||||
|
Markers (E PCR) |
Total DNA, b.p. |
189189453 |
|||||
|
Total |
3908 |
Total RNA, b.p. |
1376199 |
||||
|
assigned to other chromosomes |
880 |
Running time, approx |
3 days |
||||
|
Unassigned |
90 |
||||||
Application 2: System for finding alternative splicing
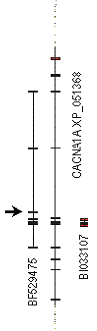
This system starts by downloading the RNA sequence with a given accession number and submitting it to BLAST search in the EST database. All hits are aligned against the given genome region using a local BLAST program generating a CorelDraw script to plot these alignments. This tool was used for searching the potentially unknown splicing variants of the neuronal calcium channel gene CACNA1A. The corresponding protein has alternatively spliced forms. Some of these forms are described in 14 related NCBI database records. In human EST database, we found two previously undefined sequences that can be completely aligned against different regions of the human chromosome 19p region to which this gene is localized (contig NT_011247). The two sequences have no reliable hits to the other genome regions and must therefore represent additional splicing variants of this gene (Figure 3). Particularly interesting is the sequence BF529475, placed into the database by National Institutes of Health, Mammalian Gene Collection (MGC). This sequence, isolated from human brain anaplastic oligodendroglioma with 1p/19q loss, shows one additional exon, typically not present in the RNA for this protein, that creates a pre-mature stop codon after 3/4 of the coding sequence. It is difficult to conclude something without experimental verification, but normal cells might have some additional mechanism for excluding this exon, and the regulating genes may be expected at 1p or 19q. An alternative explanation would be that these genes are suppressed in this tumor cell line. It may be even possible that the found truncated form is expressed under certain transient or very specific conditions physiologically.
The other sequence, BI033107 (Dias et al, 2000), from the normal nervous, includes a typical exon of CACNA1A, flanked by two typically non-coding DNA regions corresponding to the genomic region. This way of splicing creates a stop codon quite near to the truncation area of the first example however, this sequence is too short to determine the most likely reading frame which may not be identical to that of the normally spliced protein. Similar cases of alternative splicing leading to truncated cation channel proteins have been reported in sodium channels (Plummer et al., 1997 ).
|
Fig 1. Putative exon in BF529475 and alternative exons in BI033107, as they are seen in the local alignment of these RNA sequences against the chromosome 19p region encoding CACNA1A gene.The new exon in BF529475 is marked by arrow. |
Application 3: Splicing signals analyzer
Sight was used to look at the average distribution of nucleotide frequencies at the intron/exon boundaries and around the intronic splicing branch point of gemonic DNA. Our system consisted of a sequence retriever, GenScan module and specialized analyzers. We plotted the occurrence, relative frequencies, and levels of confidence (Figure 4). Apart from the known pyrimidine-rich region downstream of the branch point (Keller, Noon, 1984 ), we found a significant tendency for another pyrimidine-rich region upstream of the branch point, and confirmed the known information about the pyrimdine-rich region near the splicing acceptor site. In the coding regions, the system detected a significant decrease of T in the first position of each coding triplet. As all stop codons start with T, some evolutionary adaptation may exist to avoid combinations where a mutation is likely to create premature stop codon.
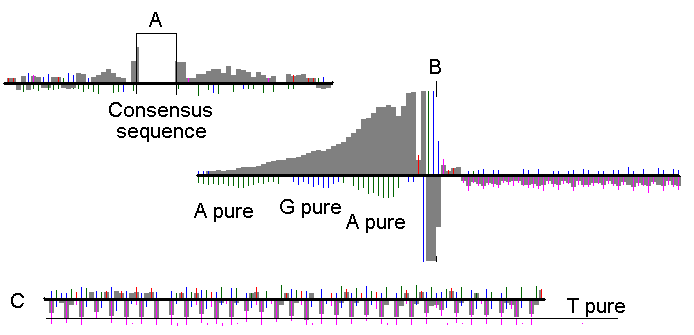
Fig 2. Statistics, representing the relative frequency of nucleotides around the branch point (A), acceptor site (B) and the coding region near to the donor site (C). The information was obtained by averaging 22463 (for A) and 130564 (for B and C) sequences from human genome as predicted by GenScan. The white rectangle in A represents the area of consensus sequence. Single vertical dash represent the relative frequency of a certain nucleotide (in this figure, the same black color is used for all). Grey bars represent the relative frequency of pyrimidines at the corresponding position. Bars and dashes upward from the horizontal black line indicate the increased frequency (with the maximal value of 5 and more times above the randomly expected frequency), bars and dashes downwards - decreased frequency (maximal value of 5 and more times below the randomly expected frequency). In this figure, only data with a confidency level of 99 % and higher were taken into consideration.
Application 4: Alternative gene prediction strategy
Instead of using GenScan or another gene prediction tool, we decided to search for sequences encoding known genes in other organisms that are not yet annotated as part of the human genome. A database was generated by selecting all proteins containing known ion channel signatures from NCBI non redundant protein database. Corresponding DNA sequences were then submitted to NCBI BLAST search against the NCBI non redundant nucleic acid database. This search returned hits to the different ESTs, annotated RNA sequences, and genome regions. Next, it was checked whether the hit with the highest similarity level points to a sequence from the organism other than Homo sapiens. In this case, it was added to the report.
We scanned the ion channel genes on chromosome 8 and the program reported two sets of fragments. Both sequences were not mentioned in the corresponding NCBI Center contig and were more similar to the sequences from organisms other than human. For each of these fragments, all received BLAST hits (E< 10-15) were exclusively hits to the different ion channel sequences. In clones AP000075.1 and adjacent AP000074.1 of contig NT_008251.5, the program detected three unknown fragments, that can be clearly identified as potential parts of the potassium large conductance pH-sensitive channel (Figure 5). All three fragments are most similar (E= 2 10-23 , 4 10-29 and 10-21) to the different sections of the same potassium large conductance pH-sensitive channel (subfamily M, alpha member 3, (gi:6680542) from mouse. They are also more remotely similar to the human maxi K channel ((Meera et al., 1996 ), gi:1588671) in the pore-forming domain (IPR001622 in InterPro).

Fig 3. Multiple alignments of the potentially coding sequence found in the clones AP000075.1 and adjacent AP000074.1, against the most similar protein (Mus musculus potassium large conductance pH-sensitive channel, subfamily M, alpha member 3, gi:6680542) (mouse) and the most similar human protein (Homo sapiens human maxi K channel). Identities are highlighted.
Another 4 potentially coding fragments (Figure 6) were detected in the clone AC067890.3 of contig NT_023670.3. All fragments were most similar (E< 5´ 10-23) to different sections of the Rattus norvegicus neuronal erg 2 potassium channel (Shi et al., 1997 ), gi:2745728) but more remotely to the Homo sapiens Eag-related gene member 2 (cyclic nucleotide gated potassium channel, Titus & Ganetzky, 2000, unpublished results, gi:13540548). The fragments represent non-transmembraneous parts of the protein (N- and C- terminal sequences). They contain PAS (IPR000014) and partially PAC (IPR001610) domains that are found in this systematic group of ion channels (Zhulin et al., 1997 ). One of the fragments also contains a cyclic nucleotide binding domain signature (IPR000595). Hence, it is also possible to think that we are observing fragments of an evolutionary old ion channel gene. However, from the version Ac067890.4, the sequencing group excluded a large region of this clone covering the 4 fragments even though they were not part of the yeast or bacterial genome nor part of the cloning and sequencing vectors. We must therefore leave the question about the presence of this sequence in the human genome open at this time.

Fig 7. Multiple alignments of the potentially coding sequence, found in the clone AC067890.3 (X) against the most similar protein (Rattus norvegicus neuronal erg 2 potassium channel, gi:2745728) (Rat) and the most similar human protein (Homo sapiens Eag-related gene member 2, gi:13540548). Identities are highlighted.
Perspective
The method described in this paper can save hundreds of hours of time for qualified researchers. It is quite resistant to the server or network errors as long as they are not persistent. The problems caused by the inappropriate behavior of the internet robots are minimized by the Sight organizing and security environment. The large number of successful applications indicates that the suggested method has good prospects. The popularity of internet robots will increase if a set of specialized code generators as introduced here become available because then the know-how required by the programmer to implement a special task is greatly reduced. When this happens, it is to be expected that several internet sites may provide internet servers just for robots that offer multiple possibilities to access data or to communicate bilaterally. Connections must then be organized more efficiently with periods of "time out" e.g. when the robots are analyzing the received data. Currently, a few such solutions exists in NCBI BLAST or InterPro server systems where the request identifier is sent to the user after submission and the results, when ready, are accessed using this identifier.
Acknowledgements
This work was supported by the Interdisciplinary Clinical Research Center (iZKF) of Ulm University funded by the General Ministry of Research (BMBF) and the GRU 460 Graduate College funded by the German Research Foundation (DFG).